

100 mg
For the use of a Registered Medical Practitioner or a Hospital or a Institution only.
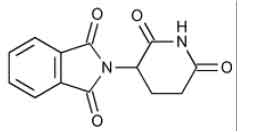
THALIDOMIDE CAPSULES USP (Thalidomide) is a immunomodulator. Chemically Thalidomide is 1H -Isoindole-1,3(2H )-dione, 2-(2,6-dioxo-3-piperidinyl)-, (±)-. The molecular formula is C13H10N2O4 and molecular weight is 258.23.
STRUCTURAL FORMULA :
Its structural formula is :
THALIDOMIDE CAPSULES USP are hard gelatin capsules containing off-white to white coloured powder.
COMPOSITION :
Each hard gelatin capsule contains :
Thalidomide USP 100 mg
Excipients q.s.
Approved colours used in empty capsule shells.
ACTIONS :
Thalidomide is an immunomodulatory agent with a spectrum of activity that is not fully characterized. In patients with erythema nodosum leprosum (ENL) the mechanism of action is not fully understood. Available data from in vitro studies and preliminary clinical trials suggest that the immunologic effects of this compound can vary substantially under different conditions, but may be related to suppression of excessive tumour necrosis factor-alpha (TNF-(alpha) production and down-modulation of selected cell surface adhesion molecules involved in leukocyte migration. For example, administration of thalidomide has been reported to decrease circulating levels of TNF-(alpha) in patients with ENL, however, it has also been shown to increase plasma TNF-(alpha) levels in HIV-seropositive patients.
PHARMACOKINETICS :
Absorption :
Absorption of thalidomide is slow after oral administration. The maximum plasma concentrations are reached 1-5 hours after administration. Co-administration of food delayed absorption but did not alter the overall extent of absorption.
Distribution :
The plasma protein binding of the (+)-(R) and (-)-(S) enantiomers was found to be 55 % and 65 % respectively. Thalidomide is present in the semen of male patients at levels similar to plasma concentrations. Therefore, because of the known severe teratogenic effects of the product, during treatment with thalidomide and for 1 week after stopping the treatment, male patients must use condoms if their partner is pregnant or is of childbearing potential not using effective contraception. The distribution of thalidomide is not influenced by age, gender, renal function and blood chemistry variables, to any significant level.
Biotransformation :
Thalidomide is metabolised almost exclusively by non-enzymatic hydrolysis. In plasma, unchanged thalidomide represents 80 % of the circulatory components. Unchanged thalidomide was a minor component (< 3 % of the dose) in urine. In addition to thalidomide, hydrolytic products N-(o- carboxybenzoyl) glutarimide and phthaloyl isoglutamine formed via non-enzymatic processes are also present in plasma and in majority in urine. Oxidative metabolism does not contribute significantly to the overall metabolism of thalidomide. There is minimal cytochrome P450 catalysed hepatic metabolism of thalidomide. There are in vitro data indicating that prednisone may give rise to enzyme induction which could reduce the systemic exposure of concomitantly used medicinal products. The in vivo relevance of these findings is unknown.
Elimination :
The mean elimination half-life of thalidomide in plasma following single oral doses between 50 mg and 400 mg was 5.5 to 7.3 hours. Following a single oral dose of 400 mg of radio-labelled thalidomide, the total mean recovery was 93.6 % of the administered dose by Day 8. The majority of the radioactive dose was excreted within 48 hour following dose administration. The major route of excretion was via the urine (> 90 %) while faecal excretion was minor. There is a linear relationship between body weight and estimated thalidomide clearance; in multiple myeloma patients with body weight from 47 - 133 kg, thalidomide clearance ranged from approximately 6 - 12 l/h, representing an increase in thalidomide clearance of 0.621 l/h per 10 kg body weight increase.
Linearity/non linearity :
Total systemic exposure (AUC) is proportional to dose at single-dose conditions. No time dependency of the pharmacokinetics has been observed.
Hepatic and renal impairment :
The extent of thalidomide metabolism by the liver cytochrome P450 system is minimal and intact thalidomide is not excreted by the kidney. Measures of renal function (CLcr) and liver function (blood chemistry) indicate minimal effect of kidney and liver function on the pharmacokinetics of thalidomide.
The pharmacokinetics of thalidomide in patients with renal dysfunction have not been determined.
Administration :
THALIDOMIDE CAPSULES USP are for oral administration.
Method of Administration :
THALIDOMIDE CAPSULES USP should be taken as a single dose at bedtime, to reduce the impact of somnolence. This medicinal product can be taken with or without food. Thalidomide treatment must be initiated and monitored under the supervision of physicians with expertise in managing immunomodulatory or chemotherapeutic agents and a full understanding of the risks of thalidomide therapy and monitoring requirements.
Dosage :
The recommended oral dose is 200 mg per day. A maximum number of 12 cycles of 6 weeks should be used. Patients should be monitored for : thromboembolic events, peripheral neuropathy, rash/skin reactions, bradycardia, syncope, somnolence, neutropenia and thrombocytopenia. Dose delay, reduction or discontinuation, dependent upon the NCI CTC (National Cancer Institute Common Toxicity Criteria) grade, may be necessary. Thromboembolic events Thromboprophylaxis should be administered for at least the first 5 months of treatment especially in patients with additional thrombotic risk factors. Prophylactic antithrombotic medicinal products, such as low molecular weight heparins or warfarin, should be recommended. The decision to take antithrombotic prophylactic measures should be made after careful assessment of an individual patient’s underlying risk factors. If the patient experiences any thromboembolic events, treatment must be discontinued and standard anticoagulation therapy started. Once the patient has been stabilised on the anticoagulation treatment and any complications of the thromboembolic event have been managed, the thalidomide treatment may be restarted at the original dose dependent upon a benefit-risk assessment. The patient should continue anticoagulation therapy during the course of thalidomide treatment.
Neutropenia
White blood cell count and differential should be monitored on an ongoing basis, in accordance with oncology guidelines especially in patients who may be more prone to neutropenia. Dose delay, reduction or discontinuation, dependent upon the NCI CTC grade, may be necessary.
Thrombocytopenia
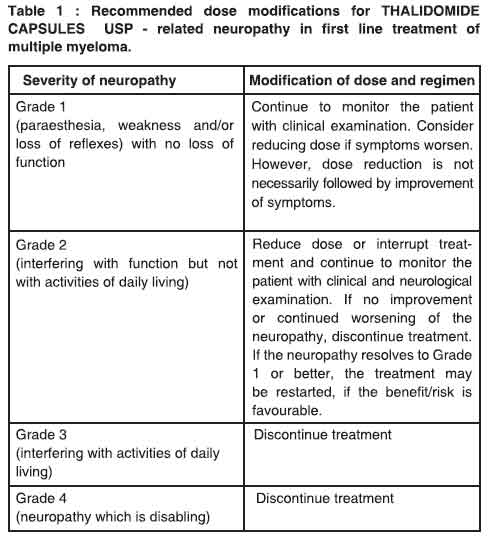
Platelet counts should be monitored on an ongoing basis, in accordance with oncology guidelines. Dose delay, reduction or discontinuation, dependent upon the NCI CTC grade, may be necessary.Peripheral neuropathy Dose modifications due to peripheral neuropathy are described in Table 1.
Elderly population
No specific dose adjustments are recommended for the elderly.
Patients with renal or hepatic impairment
THALIDOMIDE CAPSULES USP has not formally been studied in patients with impaired renal or hepatic function. No specific dose recommendations for these patient populations are available. Patients with severe organ impairment should be carefully monitored for adverse reactions.
Paediatric population
There is no relevant use of THALIDOMIDE CAPSULES USP in the paediatric population in the indication of multiple myeloma.
CONTRAINDICATIONS :
• Hypersensitivity to thalidomide or to any of the excipients.
• Pregnant women.
• Women of childbearing potential unless all the conditions of the THALIDOMIDE CAPSULES USP Pregnancy Prevention Programme are met.
• Patients unable to follow or comply with the required contraceptive measures.
WARNING :
Birth Defects :
Thalidomide can cause severe birth defects in humans. Patients should be instructed to take thalidomide only as prescribed and not to share their thalidomide with anyone else. Because thalidomide is present in the semen of patients receiving the drug, males receiving thalidomide must always use a latex condom during any sexual contact with women of childbearing potential. The risk to the foetus from the semen of male patients taking thalidomide is unknown.
Thrombotic Events :
The use of THALIDOMIDE CAPSULES USP in multiple myeloma results in an increased risk of venous thromboembolic events, such as deep venous thrombosis and pulmonary embolus. This risk increases significantly when thalidomide is used in combination with standard chemotherapeutic agents including dexamethasone. In one controlled trial, the rate of venous thromboembolic events was 22.5 % in patients receiving thalidomide in combination with dexamethasone compared to 4.9 % in patients receiving dexamethasone alone (p = 0.002). Patients and physicians are advised to be observant for the signs and symptoms of thromboembolism. Patients should be instructed to seek medical care if they develop symptoms such as shortness of breath, chest pain, or arm or leg swelling. Preliminary data suggest that patients who are appropriate candidates may benefit from concurrent prophylactic anticoagulation or aspirin treatment.
Drowsiness and Somnolence :
Thalidomide frequently causes drowsiness and somnolence. Patients should be instructed to avoid situations where drowsiness may be a problem and not to take other medications that may cause drowsiness without adequate medical advice. Patients should be advised as to the possible impairment of mental and/or physical abilities required for the performance of hazardous tasks, such as driving a car or operating other complex or dangerous machinery.
Peripheral Neuropathy :
Thalidomide is known to cause nerve damage that may be permanent. Peripheral neuropathy is a common, potentially severe, side effect of treatment with thalidomide that may be irreversible. Peripheral neuropathy generally occurs following chronic use over a period of
months; however, reports following relatively short term use also exist. The correlation with cumulative dose is unclear. Symptoms may occur some time after thalidomide treatment has been stopped and may resolve slowly or not at all. Few reports of neuropathy have arisen in the treatment of ENL despite long-term thalidomide treatment. However, the inability clinically to differentiate thalidomide neuropathy from the neuropathy often seen in Hansen’s disease makes it difficult to determine accurately the incidence of thalidomide-related neuropathy in ENL patients treated with thalidomide.
Patients should be examined at monthly intervals for the first 3 months of thalidomide therapy to enable the clinician to detect early signs of neuropathy, which include numbness, tingling or pain in the hands and feet. Patients should be evaluated periodically thereafter during treatment. Patients should be regularly counselled, questioned, and evaluated for signs or symptoms of peripheral neuropathy. Consideration should be given to electrophysiological testing, consisting of measurement of sensory nerve action potential (SNAP) amplitudes at baseline and thereafter every 6 months in an effort to detect asymptomatic neuropathy. If symptoms of drug-induced neuropathy develop, thalidomide should be discontinued immediately to limit further damage, if clinically appropriate. Usually, treatment with thalidomide should only be reinitiated if the neuropathy returns to baseline status. Medications known to be associated with neuropathy should be used with caution in patients receiving thalidomide.
Dizziness and Orthostatic Hypotension :
Patients should also be advised that thalidomide may cause dizziness and orthostatic hypotension and that, therefore, they should sit upright for a few minutes prior to standing up from a recumbent position.
Neutropenia :
Decreased white blood cell counts, including neutropenia, have been reported in association with the clinical use of thalidomide. Treatment should not be initiated with an absolute neutrophil count (ANC) of < 750/mm3. White blood cell count and differential should be monitored on an ongoing basis, especially in patients who may be more prone to neutropenia, such as patients who are HIV-seropositive. If ANC decreases to below 750/mm3 while on treatment, the patient’s medication regimen should be re-evaluated and, if the neutropenia persists, consideration should be given to withholding thalidomide if clinically appropriate.
Increased HIV Viral Load :
In a randomized, placebo controlled trial of thalidomide in an HIV-seropositive patient population, plasma HIV RNA levels were found to increase (median change = 0.42 log10 copies HIV RNA/ml, p = 0.04 compared to placebo). A similar trend was observed in a second,
unpublished study conducted in patients who were HIV seropositive. The clinical significance of this increase is unknown. Both studies were conducted prior to availability [of highly active antiretroviral therapy. Until the clinical significance of this finding is further understood, in HIV-seropositive patients, viral load should be measured after the first and third months of treatment and every 3 months thereafter.
PRECAUTIONS :
General :
The only type of thalidomide exposure known to result in drug associated birth defects are as a result of direct oral ingestion of thalidomide. Currently no specific data are available regarding the cutaneous absorption or inhalation of thalidomide in women of childbearing potential and whether these exposures may result in any birth defects. Patients should be instructed to not extensively handle or open THALIDOMIDE CAPSULES USP and to maintain storage of capsules in strip packs until ingestion. If there is contact with non-intact thalidomide capsules or the powder contents, the exposed area should be washed with soap and water. Thalidomide has been shown to be present in the serum and semen of patients receiving thalidomide. If healthcare providers or other care givers are exposed to body fluids from patients receiving THALIDOMIDE CAPSULES USP appropriate precautions should be utilized, such as wearing gloves to prevent the potential cutaneous exposure to THALIDOMIDE CAPSULES USP or the exposed area should be washed with soap and water.
Hypersensitivity :
Hypersensitivity to THALIDOMIDE CAPSULES USP has been reported. Signs and symptoms have included the occurrence of erythematous macular rash, possibly associated with fever, tachycardia, and hypotension, and if severe, may necessitate interruption of therapy. If the reaction recurs when dosing is resumed, should be discontinued.
Bradycardia :
Bradycardia in association with thalidomide use has been reported. Cases of bradycardia have been reported, some required medical interventions. The clinical significance and underlying etiology of the bradycardia noted in some thalidomide-treated patients are presently unknown.
Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis :
Serious dermatologic reactions including Stevens-Johnson syndrome and toxic epidermal necrolysis, which may be fatal, have been reported. THALIDOMIDE CAPSULES USP should be discontinued if a skin rash occurs and only resumed following appropriate clinical evaluation. If the rash is exfoliative, purpuric, or bullous or if Stevens-Johnson syndrome or toxic epidermal necrolysis is suspected, use of THALIDOMIDE CAPSULES USP should not be resumed.Seizures : Although not reported from pre-marketing controlled clinical trials, seizures, including grand mal convulsions, have been reported during post-approval use of Thalidomide in clinical practice. Because these events are reported voluntarily from a population of unknown size, estimates of frequency cannot be made. Most patients had disorders that may have predisposed them to seizure activity, and it is not currently known whether thalidomide has any epileptogenic influence. During therapy with thalidomide, patients with a history of seizures or with other risk factors for the development of seizures should be monitored closely for clinical changes that could precipitate acute seizure activity.
Pregnancy : Category X
Because of the known human teratogenicity of thalidomide, thalidomide is contraindicated in women who are or may become pregnant and who are not using the two required types of birth control or who are not continually abstaining from heterosexual sexual contact. If thalidomide is taken during pregnancy, it can cause severe birth defects or death to an unborn baby. Thalidomide should never be used by women who are pregnant or who could become pregnant while taking the drug. Even a single dose [1 capsule (regardless of strength)] taken by a pregnant woman can cause birth defects. If pregnancy does occur during treatment, the drug should be immediately discontinued. Under these conditions, the patient should be referred to an obstetrician/gynaecologist experienced in reproductive
toxicity for further evaluation and counselling. Because thalidomide is present in the semen of patients receiving the drug, males receiving thalidomide must always use a latex condom during any sexual contact with women of childbearing potential. The risk to the foetus from the semen of male patients taking thalidomide is unknown.
A pre- and postnatal reproductive toxicity study was conducted in pregnant female rabbits. Compound-related increased abortion incidences and elevated foetotoxicity were observed at the lowest oral dose level of 30 mg/kg/day (approximately 1.5 - fold the maximum human dose based upon BSA) and all higher dose levels. Neonatal mortality was elevated at oral dose levels to the lactating female rabbits ≥ 150 mg/kg/day (approximately 7.5 - fold the maximum human dose based upon BSA). No delay in postnatal development, including learning and memory functions, were noted at the oral dose level to the lactating female rabbits of 150 mg/kg/day (average thalidomide concentrations in milk ranged from 22 to 36 μg/ml).
Nursing Mothers :
It is not known whether thalidomide is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from thalidomide, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Paediatric Use :
Safety and effectiveness in paediatric patients below the age of 12 years have not been established.
SIDE EFFECTS :
Thalidomide may cause peripheral neuropathy, characterized by axonal degeneration without demyelination, affecting mainly sensory fibers in the lower limbs. Neuropathy is manifested initially by paresthesia of the feet, then the hands, followed by burning sensations in the extremities and by muscle cramps. Distribution is generally in a stocking-glove pattern. Thalidomide does not cause major motor disability, although distal weakness in the feet and depression of the ankle deep tendon reflexes do occur late in the course of the neuropathy; on discontinuation, motor signs revert more readily and completely than sensory symptoms. Patients with mild, new onset, or progressive symptoms of peripheral neuropathy should discontinue thalidomide treatment. Thalidomide-induced neuropathy progresses gradually, over a period of weeks to months; neuropathy generally is reversible if thalidomide treatment is stopped in the early stages of neuropathy. However, symptoms may occur some time after thalidomide treatment has been stopped. Although some researchers have suggested a relationship between the total dose given and the neuropathy, others found no statistically significant correlation between the severity of this effect and total dose.
However, the risk of developing poly neuropathy may be 10 % or higher in chronically treated patients who do not have Hansen’s disease. Results from one clinical study have suggested that smoking may have a protective effect against the development of peripheral neuropathy. Electrophysiological abnormalities detected before the onset of subjective symptoms have been reported. The most prominent electrophysiological alteration was a decreased sensory nerve action potential (SNAP) amplitude, but decreased sensory and motor conduction velocities, as well as alterations in latencies, were also observed. The number of patients found to have developed neuropathy was consistently higher if electrophysiological tests, rather than clinical symptoms alone, were used for diagnosis. The reduced or absent sensitivity in the extremities was frequently irreversible or was only partially reversible over a long period. The incidence of peripheral neuropathy has ranged widely, from 0.5 to 50 %. In human immunodeficiency virus (HIV)-infected patients, the incidence reportedly varies from 15 to 50 %. Patients with pre-existing neuropathies may be more sensitive to the development of thalidomide-induced neuropathy. However, neuropathy may also be seen in HIV disease itself. The side effects of thalidomide appear to be more severe and more poorly tolerated in HIV-infected patients. Detecting thalidomide-induced neuropathy in erythema nodosum leprosum (ENL) patients may be difficult because of the similarity of clinical symptoms and changes in electrophysiological measurements that result from the underlying ENL disease. The incidence of peripheral neuropathy due to thalidomide appears to be low in patients being treated for ENL; this may be because peripheral nerves are consistently affected in lepromatous leprosy and thalidomide improves the neuritis of ENL by reducing inflammation.
Gradual dose escalation may be efficacious in avoiding adverse reactions. The following side/adverse effects have been selected on the basis of their potential clinical significance.Incidence more frequent Peripheral neuropathy tingling, burning, numbness, or pain in the hands, arms, feet, or legs; muscle weakness. Note : If symptoms of thalidomide-induced neuropathy develop, thalidomide should be discontinued immediately to limit further damage. Usually, treatment with thalidomide should be reinitiated only if the neuropathy returns to baseline status.
Patients should be instructed to not extensively handle or open THALIDOMIDE CAPSULES USP and to maintain storage of capsules in strip packs until ingestion. Patients should be instructed not to share medication with anyone else. Patients should be instructed that thalidomide frequently causes drowsiness and somnolence. Patients should be instructed to avoid situations where drowsiness may be a problem and not to take other medications that may cause drowsiness without adequate medical advice. Patients should be advised as to the possible impairment of mental and/or physical abilities required for the performance of hazardous tasks, such as driving a car or operating other complex machinery. Patients should be instructed that thalidomide may potentiate the somnolence caused by
immediately. Patients should also be instructed that thalidomide may cause dizziness and orthostatic hypotension and that, therefore, they should sit upright for a few minutes prior to standing up from a recumbent position. Patients should be instructed that they are not permitted to donate blood while taking thalidomide. In addition, male patients should be instructed that they are not permitted to donate sperm while taking thalidomide.